BioMakie.jl
Status:

![]()
![]()
Installation
To install BioMakie, access the Julia package REPL by pressing ] from the Julia REPL, then run add BioMakie.
Contributing and questions
Anyone can contribute to this package, by doing things like reporting bugs, fixing issues, improving performance, adding new features, and adding examples. Feel free to open an Issue or Pull Request, or communicate on the #biology or #makie channels of the Julia Slack.
About
This package provides plotting functions for protein structures, multiple sequence alignments, and some other related plots like protein structure contact maps. It also provides more complicated examples that show off additional functionality and interfaces. The main plotting functions are plotstruc and plotmsa, along with their mutating versions, plotstruc! and plotmsa!.
Implemented packages:
Significant or full coverage:
- BioStructures.jl
- MIToS.jl
- FastaIO.jl
- FASTX.jl
Some coverage:
- MolecularGraph.jl
- ProtoSyn.jl
Implemented visualizations:
- Structures
- Ball and stick, spacefilling, covalent representations
- Selections
- Alpha shapes
- Multiple sequence alignments
- Grid display
- Selections
- Frequency plot
- Data acquisition and GPT-3.5-turbo API
- Mutating structural residues with ProtoSyn
To Do:
- Non-standard and modified amino acids
- Connect MSA and structure plot
- Protein dynamics
- Better support for ligands and multiple chains
- Database web API interfaces
- WGLMakie support
- More examples
using BioMakie
using GLMakie
using BioStructures
struc = retrievepdb("2vb1") |> Observable
## or
struc = read("2vb1.pdb", BioStructures.PDBFormat) |> Observable
fig = Figure()
plotstruc!(fig, struc; plottype = :ballandstick, gridposition = (1,1), atomcolors = aquacolors)
plotstruc!(fig, struc; plottype = :covalent, gridposition = (1,2))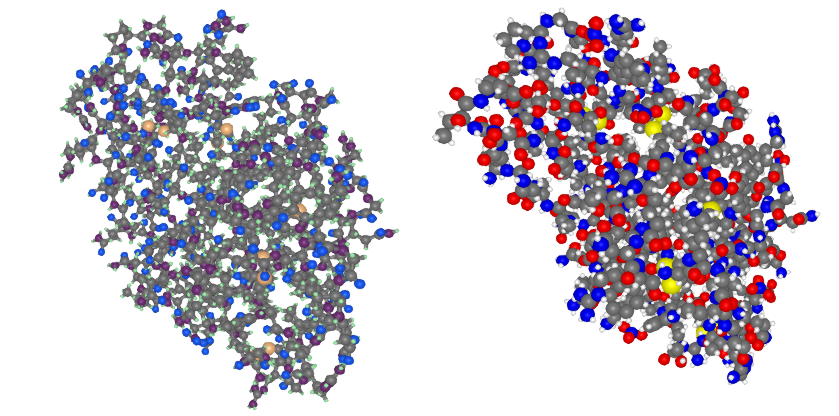
To view a multiple sequence alignment, use the plotmsa function with a Pfam MSA or fasta file.
using FASTX
reader = open(FASTX.FASTA.Reader, "PF00062_full.fasta")
msa = [reader...] |> Observable
close(reader)
## or
using MIToS.MSA
msa = read_file("pf00062.stockholm.gz", Stockholm)
fig = plotmsa(msa; colorscheme = :tableau_blue_green)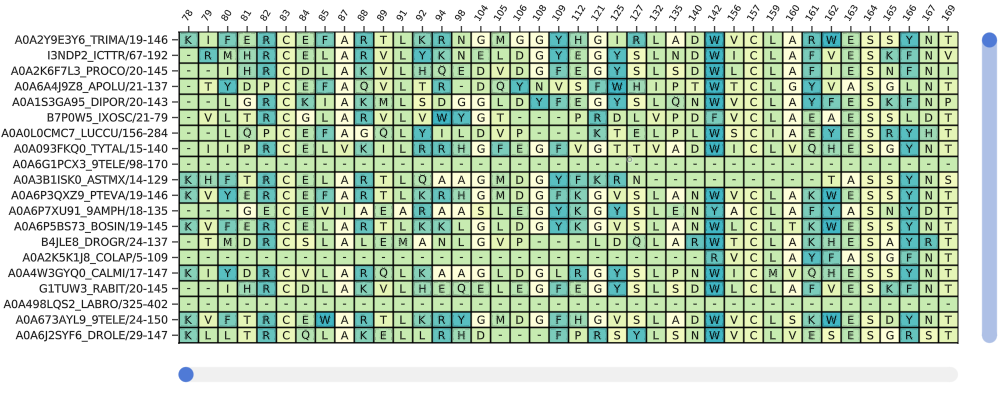
Other examples
Viewing the frequencies of amino acids in a multiple sequence alignment
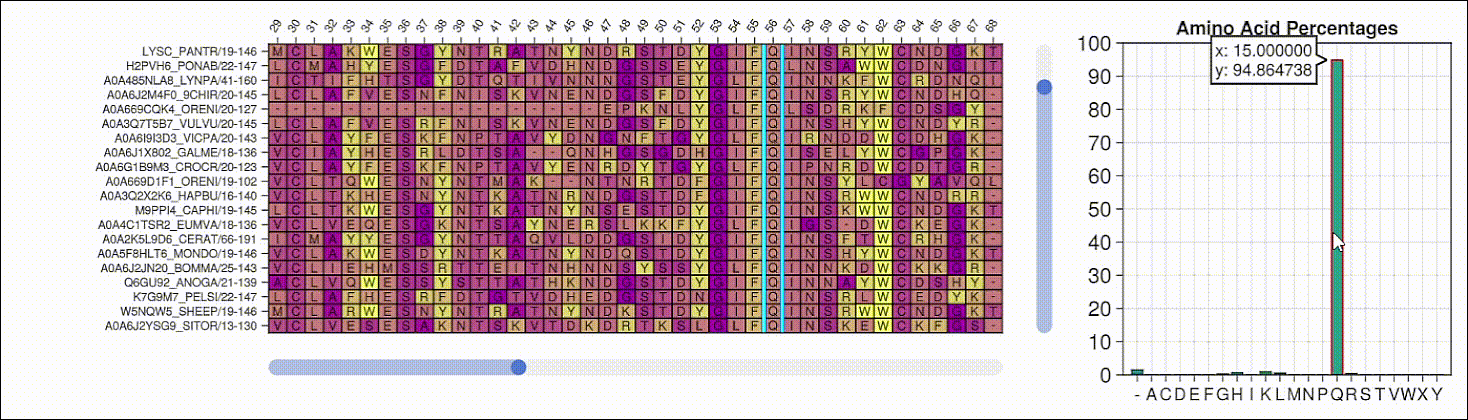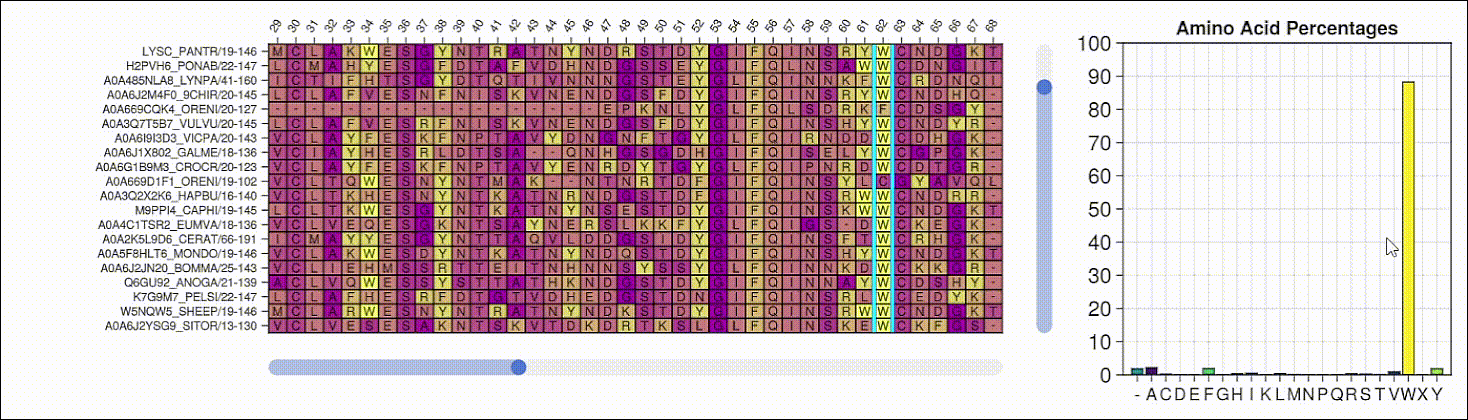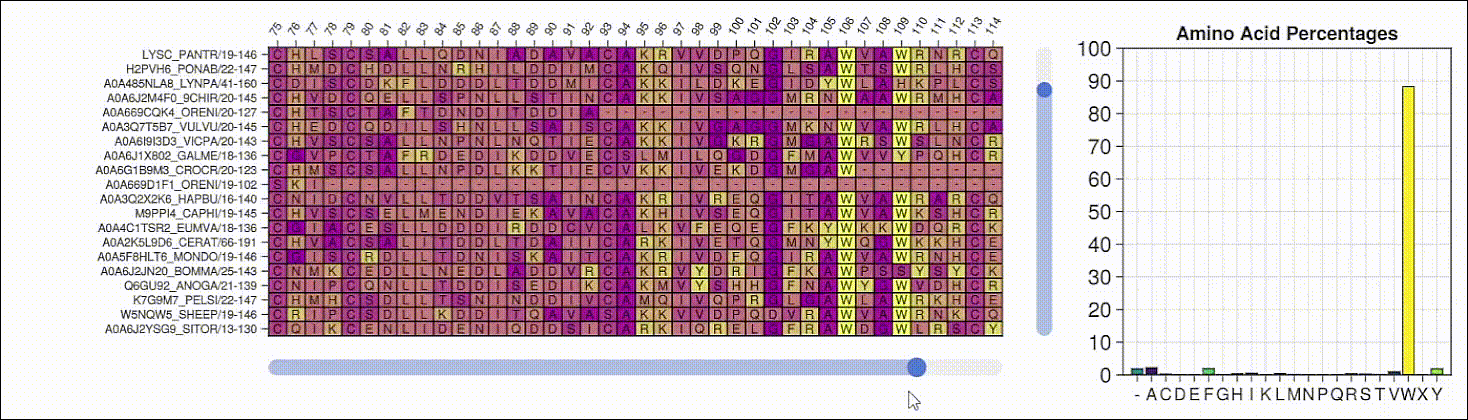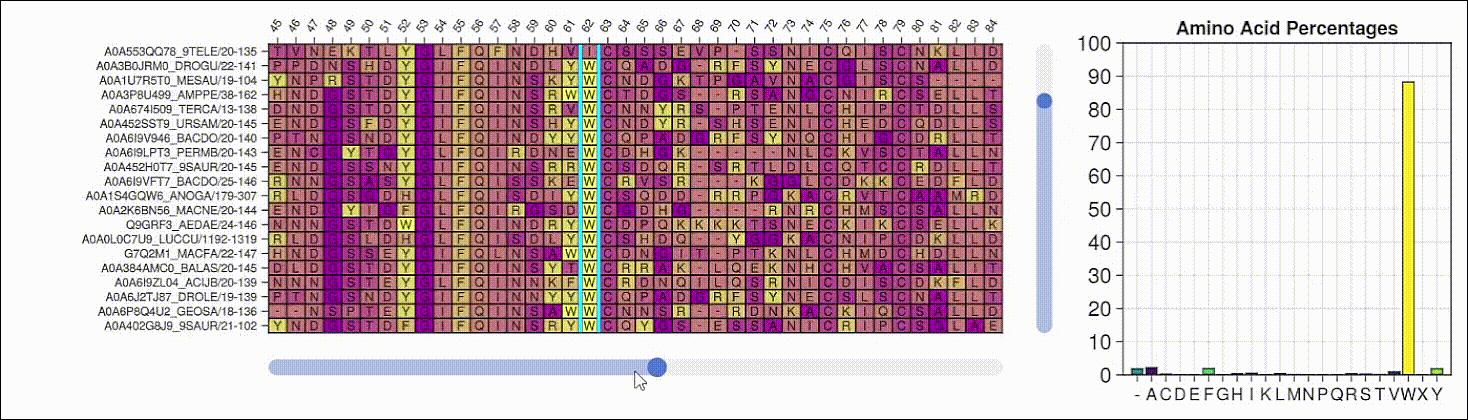
Alpha shapes can be used to visualize the surface of a protein structure
Database information can be displayed for a protein (including a GPT response, OpenAI.jl)

Protein residue mutation and changing rotamers (with ProtoSyn.jl)
